

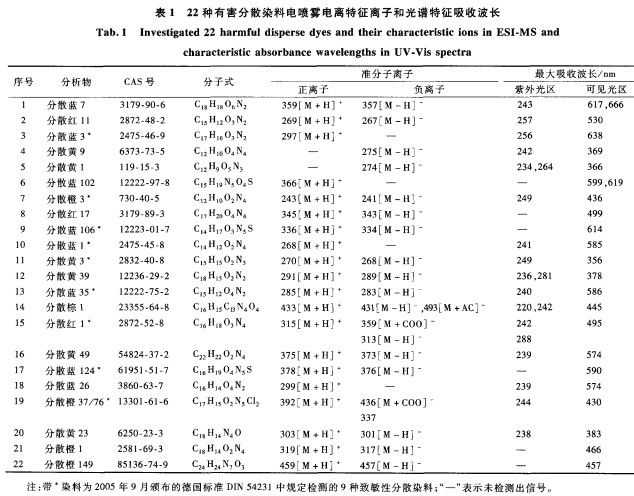
分散染料是在水溶液中呈分散状态的染料总称,主要用于聚酯、聚酰胺和醋酯等化学纤维及其混纺制品的染色。分散染料对人体皮肤的致敏性一直备受关注,20世纪7O年代报道的“锦纶丝袜过敏”事件就缘于锦纶纤维上的分散染料造成接触性皮炎川。据报道,三分之二的纺织品致敏事件由分散染料造成。目前有20种常用于纺织品染色的分散染料被确认为致敏染料,生态纺织品标签OekorexStandard100和欧盟Eco―label(EU2002/371/EC)都要求纺织品中不得检出这20种染料。由于分散黄23和分散橙149易分解生成具有致癌致敏性的对氨基偶氮苯,Oeko―texStandard100从2006年起要求纺织品中也不得检出这2种染料。分散染料常用的检测方法有薄层色谱法(TLC)、高效液相色谱法(HPLC―DAD)、液相色谱一质谱法(LC―MS)和液相色谱一串联质谱法(LC―MS/MS)。2005年9月,德国标准化协会颁布了标准检测方法DIN54231,规定用超声波辅助甲醇方法提取和液相色谱一二级管陈列检测器一质谱(LC.DAD―MS)方法检测纺织品中的9种分散染料。
分散染料为分子质量相对较小的脂溶性染料,性质相近,而且染料中常含有合成中间体、同分异构体和分散剂等杂质;故这22种分散染料的检测要求检测仪器的色谱分离性能较高,检测人员的定性鉴别能力较强。本文用1.8txm的细粒径填料色谱柱,在超高压条件下快速、高效分离22种有害分散染料,根据染料的光谱吸收特征和串联质谱的结构信息进行准确测定,同时,用氯苯蒸汽回流萃取纺织品中分散染料。
1.实验部分
1.1仪器与试剂
Accela高效液相色谱仪一Accela二极管光谱检测器一Access串联四级杆质谱LC―PDA.MS/MS(美国ThermoFisher公司),配有2IxLPDA流通池、电喷雾化学电离源(ESI),Accela高压泵的最大承受压力为8×10Pa;提取装置(自制,参见文献旋转蒸发仪(德国BUCHI公司);0.20m聚四氟乙烯薄膜过滤头(津腾公司)。
22种分散染料(见表1)有证标准物质均购自德国Dr.Ehrenstorfer公司;甲醇、乙腈(色谱纯,德国Merck公司);乙酸铵(色谱纯,美国Sigma公司)水(由MilliQ系统纯水制备)。
1.2标准工作液制备
准确称取各种染料的标准物质(精确至0.01ms),用甲醇为溶剂准确配制得1.0g/L贮备液,于一18℃避光保存。以甲醇为溶剂,对贮备液进行梯级稀释,配制22种染料的混合标准工作溶液,每种染料的质量浓度为1mg/L(于4℃保存4周)。
1.3样品前处理
取代表性样品,剪成3cm×1cm的细条状,混匀。称取1.0g,精确至0.01g,用无色纱线扎紧系于提取装置的金属钩上,垂直置于20mL沸腾的氯苯上方30min。用平板电炉加热,样品被氯苯的蒸汽包围萃取染料,经过冷凝管冷却,氯苯形成回流。提取液用旋转蒸发仪去除试剂,残留物用10mL甲醇定容,用0.20lxm聚四氟乙烯薄膜过滤至样品瓶中,用LC.PDA―MS/MS分析。
1.4色谱条件
色谱柱:ZorbaxEclipseC18反相色谱柱(100mm×2.1mm,1.8m,最大承受压力为6×10Pa),带有1.8m保护柱(均为美国Agilent公司),柱温为40℃;流动相:甲醇和乙腈的混合溶剂
(体积比为40:60)(A)和10mmol/L乙酸铵溶液(B)。梯度洗脱:初始7min内A从10%线性增至55%,8~9min内再增至95%并保持2.5min,A在12min变为5%,并保持至15min;流速为300~L/min,进样量为5L。
l.5光谱条件
扫描速率为40Hz,采集波长为200~700nm,检测波长为420、570、640nlTl。样品经色谱柱分离后流入PDA光谱检测器,再直接导人带有电喷雾离子源(ESI)接口的质谱仪。
1.6质谱条件
喷雾电压:4.5kV(ESI+),3.5kV(ESI一);鞘气:N(纯度为99.99%),16L/min;辅助气:N(纯度为99.99%),4L/min;碰撞气:Ar(纯度为99.999%),23.79Pa;毛细管温度为350℃。扫描模式:选择反应监控(SRM),扫描离子对(母离子/子离子)及碰撞电压(见表2);离子对扫描时间为20ms,离子对切换时间为5ms,离子扫描宽度为0.01。中。本文对常规的索氏提取法进行改进,设计一种全新的氯苯回流提取方法:将样品直接悬置于提取溶剂上方,利用高温的氯苯蒸汽循环提取织物上的染料。而在索氏提取中,常用滤纸或纤维套管装载样品,本文的方法可有效避免染料对滤纸或纤维套管的沾色。在氯苯蒸汽回流提取实验中,染色样品化学纤维上的颜色都逐渐变浅直至无色,表明纤维上的染料被完全剥离提取下来
。
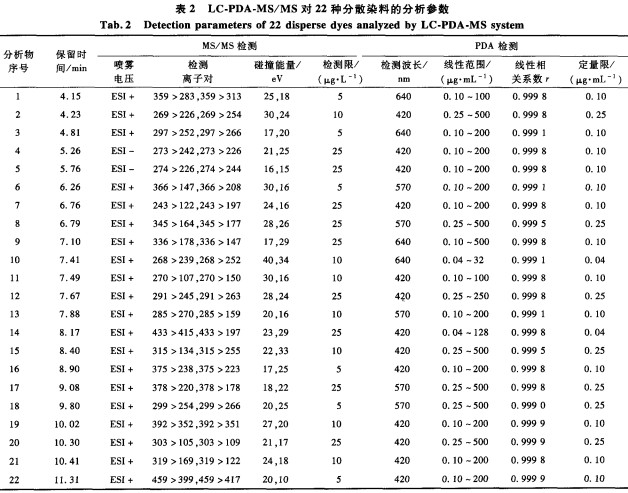
将5块阳性样品(其中2块样品都含有分散黄23)分别用氯苯蒸汽回流法和DIN54231的超声法处理,提取液用LC-PDA定量分析,并对l0次平行测试结果进行统计分析,结果见图1。图中显示,经氯苯蒸汽回流提取后,样品中分散染料的检测值为超声提取结果的2.3~11倍,表明本文方法提取效率更高。此外,本文方法的平行测试结果的相对标准偏差值(RSD)都小于10%,而超声法的RSD值较高,达17.8%~28.4%。这可能是由于在超声容器中超声能量分布不均匀所致,因此,相比较DIN54231规定的超声波辅助甲醇提取方法,本文研究的氯苯蒸汽回流提取方法更稳定,提取效率更高。
2.2色谱分离条件的选择
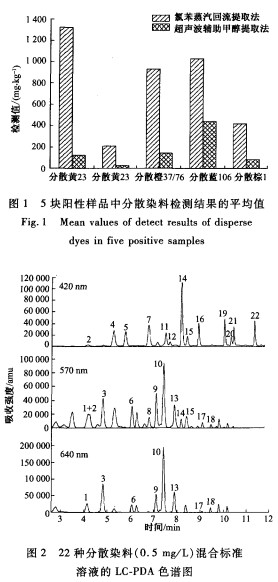
使用1.8m固定相的液相色谱柱,在超高压条件下可提供较高的分离效率,有效提高色谱分离度。图2示出了质量浓度为0.5mg/L的22种分散染料混合标准溶液的LC―PDA色谱图。可看出,除分散蓝7(1号峰)和分散红11(2号峰),其余的20种化合物都实现了基线分离。然而对于这2个共流出化合物,分散红11可在420nm下检测,分散蓝7在640nln下具有较高的检测灵敏度,而且这2种化合物在这2个波长下互不干扰,这样就可通过不同检测波长来实现这2种化合物的分离。
除提供较高的分离度,1.8I.zm色谱柱也提供了较快的分离速度,对22种染料的每次进样分析时间为15min(包括色谱柱平衡时间),而DIN5423I标准中所用的传统5txm色谱柱需用40min分离9种染料,分析时间缩短了近三分之二,大大提高了分析效率。此外,在快速分离过程中,每个分析物的峰形和色谱保留时间都未发生显著变化,显现了较稳定的分离性能。
2.3光谱检测
分析物包括8种蓝色、6种黄色、4种橙色、3种红色和1种棕色的染料。PDA检测器对每种化合物都扫描,采集得到在200~700nm范围内的紫外一可见光吸收光谱图(图略)。样品分析的色谱保留时问及其对应的光谱图,都有助于分散染料的定性分析。表2列出了每种染料在紫外光区和可见光区的特征吸收波长。为获得较好的检测灵敏度和选择性,最好在分析物的特征吸收波长处检测。由于PDA检测器最多提供3个检测波长通道,为避免紫外区域的吸收干扰,并作为妥协,选用420、570、640nm等3个可见光波长来同时检测22种染料:在420nm下检测分散黄9、1、3、39、49、23,分散橙3、37/76、1、149以及分散红11和分散棕1等l2种染料;在570nm下检测分散红17、1,分散蓝102、35、124、26等6种染料;在640nm下检测分散蓝7、3、106、1等4种染料。
2.4质谱鉴定
作为非离子型化合物,分散染料在较高的ESI喷雾电压下发生电离。经电离后,加上和减去1个质子分别形成准分子离子[M+H]和[M―H]-。表2列出了22种分散染料在ESI+和ESI一模式下电离生成的特征离子,除分散黄1、9这2种染料在正离子模式下没有响应信号而需要在ESI一模式下检测之外,其余的20种染料都在正离子模式下有较高的相应信号,用ESI+模式检测。选定一级质谱中的准分子离子作为分析物的母离子,接着进行二级质谱的碰撞裂解,得到22种分散染料的二级质谱图(图略)。根据欧盟EU657/EC对串联四级杆质谱分析要求,每个化合物选取2个离子对作为定性分析依据。
2.5分散蓝35和分散蓝26的鉴别
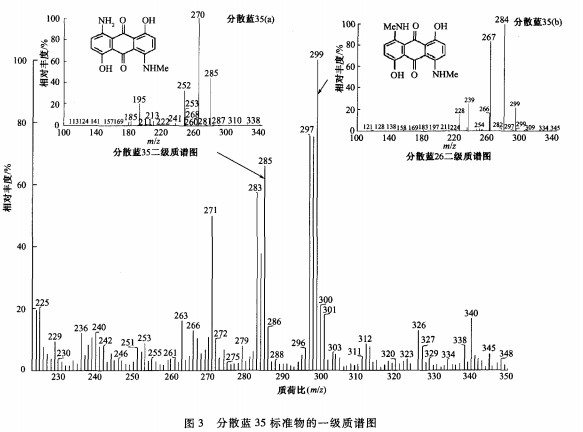
标准DIN54231中有2个化合物都为分散蓝35,分为分散蓝35(a)和分散蓝35(b)。如何鉴别和确认这2个分散蓝35是本文的重点研究内容之一。把分散蓝35的标准溶液不经过色谱分离而直接注入质谱中分析发现,其一级电离质谱中含有2个特征离子,质荷比(m/z)分别为285和299,表明分散蓝35的标准物质中可能含有其他杂质,且杂质含量较高。这2个特征离子的质量数相差14,表明这2个物质的分子质量也可能相差14,可能是相差一个一CH,一的同系物。质荷比为299的离子可能是分子质量为298的化合物[M+H]的准分子离子,而分散蓝26的分子质量恰为298。将这个离子作为母离子,在二级质谱中碰撞裂解,所获得的质谱图与分散蓝26的二级质谱图完全吻合(见图3)。此外,将分散蓝35、26的标准溶液分别用LC―PDA分析,与DIN54231描述相同的是:分散蓝35的色谱图中出现2个特征峰,虽然2个峰的光谱图相近,但是后1个色谱峰(分散蓝35(b))的保留时间与分散蓝26的保留时间一致,因此,可判断DIN54231中的分散蓝35(a)就是分散蓝35本身,分散蓝35(b)应为分散蓝26。分散蓝35和26为分子结构仅差1个亚甲基的同系物(见表1),因此,在分散蓝35的制备过程中容易产生副产物分散蓝26,而这2种物质化学性质相近,很难提纯分离,导致分散蓝35的标准物质中含有分散蓝26。
2.6检测限和线性
根据3倍信噪比(S/N=3)和10倍信噪比(S/N=10)分别计算检测限(LOD)和定量限(LOQ)。由于MS主要用于染料的定性分析,PDA主要用于染料的定量分析,本文仅计算了基于LC.MS/MS的LOD值和基于LC―PDA的LOQ值。数据表明,ESI.MS/MS的检测灵敏度要高于PDA。22种分散染料的LOQ值都较低,在0.04Ixg/mL(0.8mg/kg)~0.25txg/mL(5mg/kg)之间,都低于DIN54231中2.41txg/mL的测定低限,并低于Oeko―texStandard100规定的50mg/kg限定量。1.8m的小粒径色谱柱分离度较高,每个化合物的峰底宽不超过0.2min(见图2),与传统的5m色谱柱相比较,灵敏度至少提高了4倍。
对各种分散染料的基质匹配标准溶液和纯标准溶液所绘制的标准曲线进行比较发现,二者几乎完全重合,表明研究中由于纤维基质带来的干扰可忽略不计。表2中,所有分析物的标准曲线都表现出较好的线性(n:6,r>0.999)。
2.7回收率和精密度
图1中,5块阳性纺织品的10次平行实验结果RSD值都小于10%;向阴性涤纶织品中分别添加5、10、50mg/kg的22种分散染料进行加标回收实验,10次平行实验结果的RSD值都不大于8.4%,回收率都在86.4%~98.7%之间,且加标样品中残留物的SRM特征子离子丰度比与标准物基本一致,满足质谱方法的确证要求。
2.8样品分析
将研究建立的方法应用于实验室的日常分析,根据化合物的色谱保留时间、光谱特征吸收波长(见表1)和SRM子离子丰度比值进行鉴别。在连续2个月的进样分析期间,用这种方法对510个样品进行分析测试,Ft常维护是每天用甲醇冲洗仪器系统1次以及仅有的1次更换保护柱,这表明了该方法具有较好的稳定性和系统较强的抗干扰能力。
在分析测试的51O个样品中,有41个样品检测出致敏的分散染料,检出的有害分散染料频率是:分散黄23(15个样品)、分散橙37/76(10个样品)、分散蓝106(5个样品)、分散黄3(2个样品)、分散红1和分散橙1(都为1个样品)。对照标准曲线,对LCPDA中的色谱峰面积进行插值计算,外标法计算出阳性样品中检出染料的含量在38.2~2015mg/kg之间。检出的有害分散染料不仅存在于面料中,还存在于衬里布、裤料、帽子以及一些辅料中,如缝纫线和拉链边布。
3结论
用1.8m细粒径液相色谱柱分析法可有效提高目标物与杂质的分离效能,降低杂质的干扰。此外,依据染料的特征吸收光谱和串联质谱可提供的分子结构信息,可准确地测定纺织品中的22种有害分散染料。加标回收实验和阳性样品重复实验的结果表明,该方法回收率高,精密度好,技术指标符合分析方法要求。与德国标准DIN54231用超声波辅助甲醇提取和5m色谱柱分离测定纺织品中9种分散染料的方法相比较,氯苯蒸汽回流提取方法更稳定,提取效率更高,1.8m色谱柱的分离效率更高,分析时间更短,且检测灵敏度更高,满足生态纺织品的检测要求。
来源 丁友超,曹锡忠,蔡建和,周佳,钱凯
该文章暂时没有评论!
最新技术文章
点击排行
 安卓APP
安卓APP 微信号:yinhuashijie
微信号:yinhuashijie 手机版
手机版 《印花世界》杂志
《印花世界》杂志 微信群接入口
微信群接入口 客服
客服











